Overview
Ph+ve Acute Lymphoblastic Leukemia (Ph+ve ALL) is a clonal lymphoproliferative neoplasm, affecting B-lymphocytes. The incidence of ALL is approximately 0.8 per 100 000 adults, with 20-25% of these being Ph+ve ALL and a median age of onset of 15. Ph+ve ALL is most often characterized by the reciprocal translocation of chromosomes 9 and 22 – t(9;22)(q34.1;q11.2). The resulting derivative 22 chromosome (der22) is colloquially described as the Philadelphia chromosome (Figure 1).
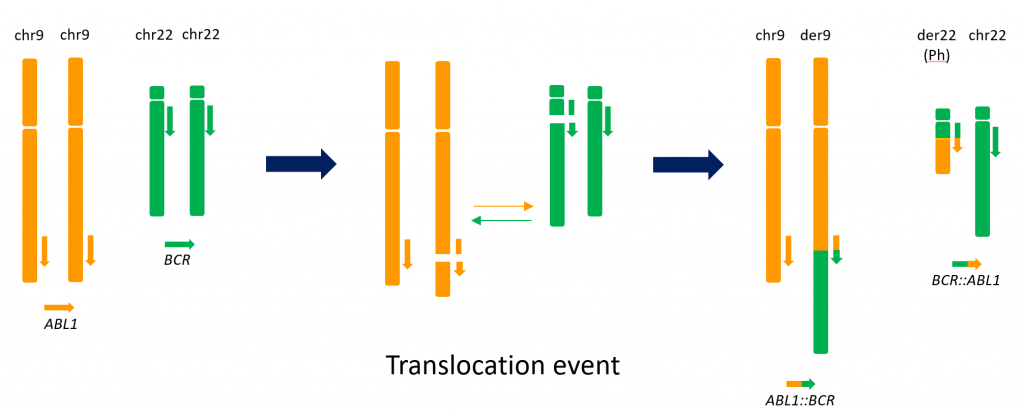
Figure 1: The reciprocal translocation of chromosomes 9 and 22 resulting in the Philadelphia chromosome
This translocation generates a novel fusion gene (BCR::ABL1) composed of the 5’ portion of the BCR gene on chromosome 22 and the 3’ portion of the ABL1 gene on chromosome 9. Several variants of the BCR::ABL1 fusion transcript are known. The most common variant associated with Ph+ve ALL (p190) fuses BCR exon 1 to ABL1 exon 2 (e1a2). More rarely, the p210 variant, fuses either BCR exon 13 or BCR exon 14 to ABL1 exon 2 (e13a2 and e14a2 respectively). Notably, the p230 variant (e19a2) is effectively absent in Ph+ve ALL.
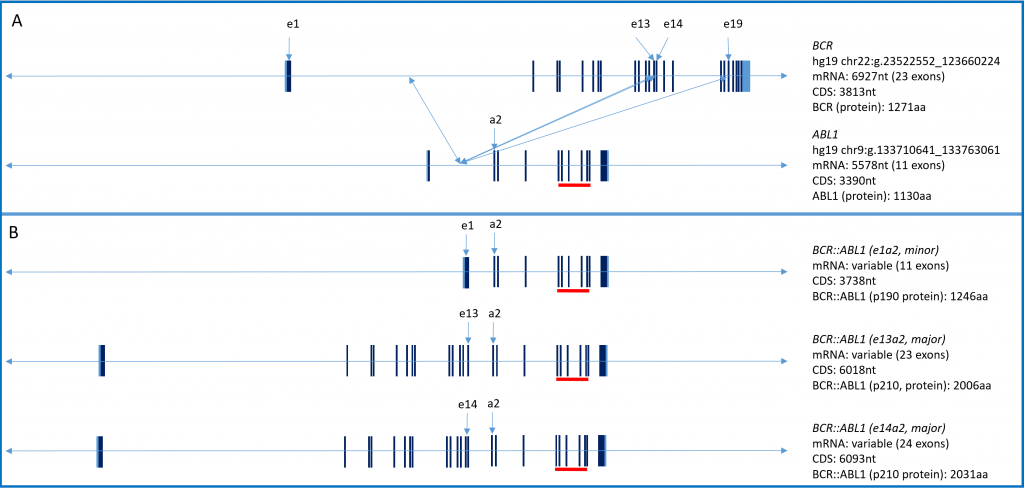
Figure 2: Panel A: Genomic organization of BCR and ABL1. Panel B: Genomic organization of the common BCR::ABL1 fusion variants in Ph+ve ALL. Red bar indicates location of the ABL1 kinase domain.
Regardless of the transcript variant present at diagnosis, it is invariably an in-frame event resulting in a novel kinase – unhindered by the regulatory domains present in the parent proteins. Expression of the fusion protein then results in the inhibitor sensitive activation of the JAK-STAT, AKT, and RAS signaling pathways.
Natural History
Prior to the advent of BCR::ABL1 targeted therapy in ~Y2K the overall survival was in the 10-20% range (patients treated primarily with intensive chemotherapy and/or stem cell/bone marrow transplant). Subsequent to the introduction of targeted therapy to the existing therapeutic regimen, the overall survival of Ph+ve ALL patients is clearly age dependent with children having the best outcome (89% at 5yrs) followed by AYA patients (61% at 5yrs) and adults (20-40% at 5yrs); with older adults stubbornly nearer the 20% mark.
Testing – Diagnostic
The laboratory diagnosis of Ph+ve ALL is currently most commonly performed by karyotype analysis of a bone marrow specimen followed by confirmation of t(9;22) by fluorescent in situ hybridization (FISH) on this very same specimen (see BCR::ABL1 FISH (Dx)).
Testing – Pre-treatment testing (Baseline)
Following the initial diagnosis of Ph+ve ALL, reflex molecular testing from peripheral blood and bone marrow derived RNA is indicated. This testing allows the laboratory to confirm that the molecular lesion present at diagnosis is amenable to molecular monitoring post initiation of treatment and by which method monitoring should proceed (see below). In the case of either a p190 or p210 transcript variant this testing also allows for the establishment of a baseline molecular burden for the patient.
Baseline testing follows the same process and method as subsequent MRD testing.
Testing – Monitoring Response to Therapy (MRD testing, draft)
Monitoring, by one of four different methods, is performed at predefined intervals depending on the treatment plan developed for the patient (see below). For e1a2(p190), e13a2(p210), or e14a2 (p210) positive disease, monitoring is by quantitative RT-PCR (QRT-PCR) and reported simply as a molecular burden. For e1a3(p190like), e13a3(p210like), e14a3(p210like), and e19a2 (p230) positive disease, monitoring is by QRT-PCR but reported simply as positive or negative for the presence of the transcript variant. For all other transcript variants, monitoring is by standard RT-PCR and is reported simply as positive or negative for the presence of the transcript variant.
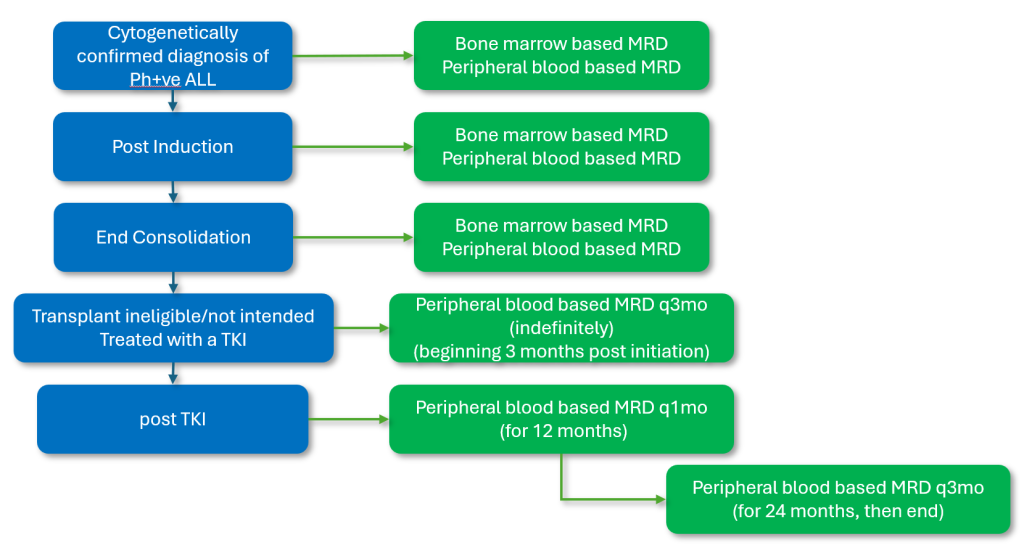
Figure 3a: Funded MRD touch points for patients for whom hematopoietic stem cell transplant is not indicated/intended.
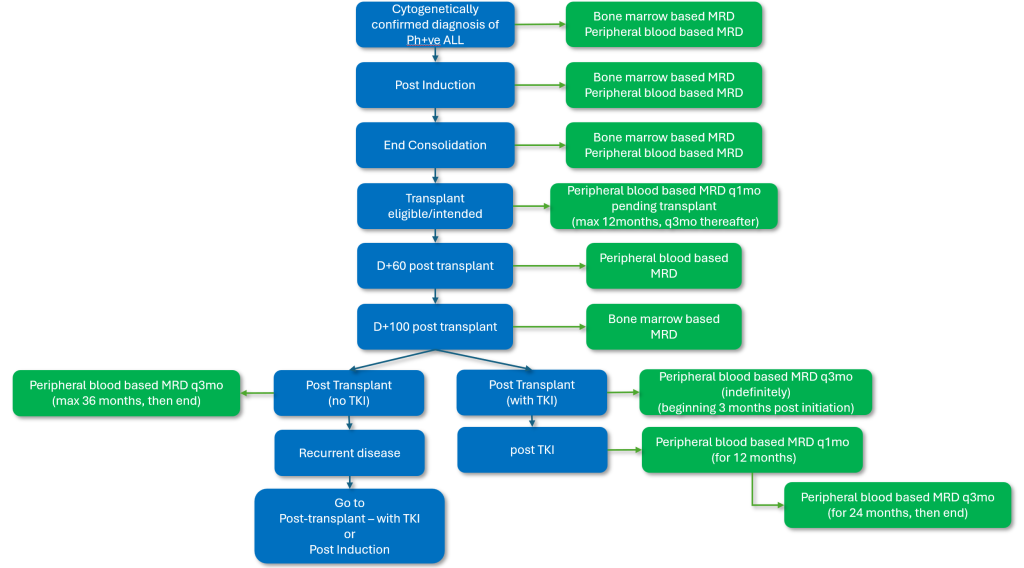
Figure 3b: Funded MRD touch points for patients for whom hematopoietic stem cell transplant is intended.
It should be noted that the above testing intervals are for routine monitoring. Testing between intervals for non-routine instances is always available (following prior consultation with the laboratory).
Testing – BCR::ABL1 Kinase Domain Mutation Screening
While response rates to treatment can be durable, it remains that patients may progress on therapy. In the event that a fully compliant patient progresses while on therapy, mutation screening of the BCR::ABL1 kinase domain may reveal the presence of a mutation. If a mutation is identified, the specific mutation may guide the choice of the next line of therapy. In general, kinase domain mutation screening is routinely informative only after a demonstrated 1 log increase in molecular burden and a molecular burden at the time of testing at, or in excess of, 1% (log burden of -2.00).
Selected References
NCCN Guidelines – Acute Lymphoblastic Leukemia v1.2022
WHO Classification of Tumours of Haematpoietic and Lymphoid Tissues (2017)